
ABSTRACT
Genetic engineering of microbes has developed rapidly along with our ability to synthesize DNA de novo. Yet, even with decreasing DNA synthesis costs there remains a need for inexpensive, rapid and reliable methods for assembling synthetic DNA into larger constructs or combinatorial libraries. While technological advances have resulted in powerful techniques for in vitro and in vivo assembly of DNA, each suffers inherent disadvantages.
Here, an ex vivo DNA cloning suite using crude cellular lysates derived from E. coli is demonstrated to amplify and assemble DNA containing small sequence homologies. Further, the advantages of an ex vivo approach are leveraged to rapidly optimize several parameters of the ex vivo DNA assembly methodology testing lysates from different engineered strains of E. coli, with various buffer components and using titrations of purified cloning enzymes.
Finally, in order to complete the cloning suite, a vector expressing the Pyrococcus furiosis (Pfu) DNA polymerase was designed, constructed and expressed in E. coli to create a ‘functionalized lysate’ capable of ex vivo PCR. Not only do we demonstrate ex vivo cloning methodology as a complete cloning package capable of replacing the expensive cloning reagents currently required by synthetic biologists, but also establish ex vivo as an overarching approach for conducting molecular biology.
EX VIVO DNA ASSEMBLY
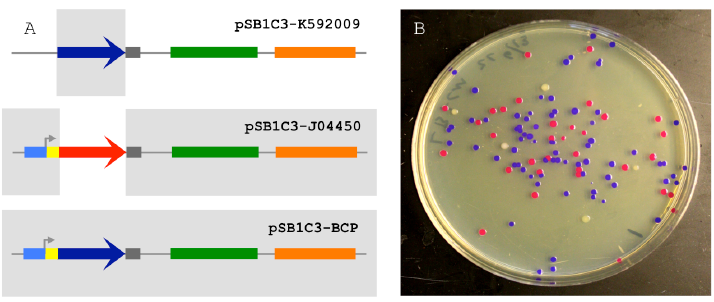
Figure 2.1 The 2-way Circular Assembly.
Figure 2.1 The 2-way Circular Assembly (A) 2-way assembly was demonstrated by joining a coding sequence for blue chromogenic protein (BCP; a 698 bp segment of pSB1C3-K592009 colored in dark blue) and the majority of pSB1C3-J04450 (a 2446 bp segment), thereby replacing the RFP coding sequence with BCP (pSB1C3-BCP). The pMB1 origin is colored in green and the chloramphenicol resistance gene is colored orange. (B) Correctly assembled plasmids allow transformants to express BCP (blue colonies) while colonies containing carryover template plasmids appear either red (pSB1C3-J04450) or white (pSB1C3-K592009).
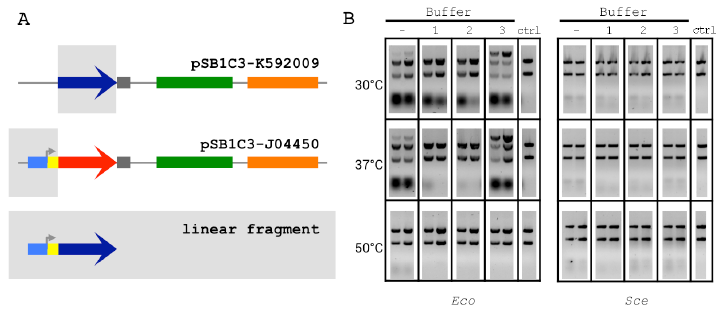
Figure 2.2 The 2-way Linear Assembly and Buffer Optimization.
Figure 2.2 The 2-way Linear Assembly and Buffer Optimization (A) Optimized ex vivo reaction conditions were identified by visualizing the joining of a 697 bp segment of pSB1C3-K592009 (BCP) and a 381 bp segment of pSB1C3-J04450 (Promoter) into a larger linear DNA molecule. (B) Reactions were run for 1 and 2 h (left and right in each gel, respectively), at three temperatures and four buffer compositions for each Eco and Sce. All buffers contained 1 mM NAD and 1 mM DTT. ATP and Mg2+ concentrations(mM:mM)varied as follows:(1)10:5,(2)5:5,(3) 1:10. The control lanes include a reaction with no supplemented buffer (−) and a negative control (“ctrl”) DNA-only lane (no buffer or lysate).

Figure 2.3 The 3-way Assembly and Colony Counts.
Figure 2.3 The 3-way Assembly and Colony Counts (A) Three-way assembly was demonstrated by joining the BCP coding sequence (a 696 bp segment of pSB1C3-K592009), the majority of a plasmid carrying a different antibiotic resistance gene (a 2214 bp segment of pSB1K3-J04450) and a promoter-RBS region used to drive BCP expression (a 387 bp segment of pSB1C3-J04450), resulting in pSB1K3-BCP.
(B,C) The pMB1 replication origin is colored in green, the chloramphenicol resistance gene is colored orange and the kanamycin resistance gene is colored purple. Colony counts for optimized two- and three-way assembly experiments. The negative control assembly reaction is labeled “None” meaning that these colonies result from in vivo assembly in E. coli.
OPTIMIZATION OF EX VIVO DNA
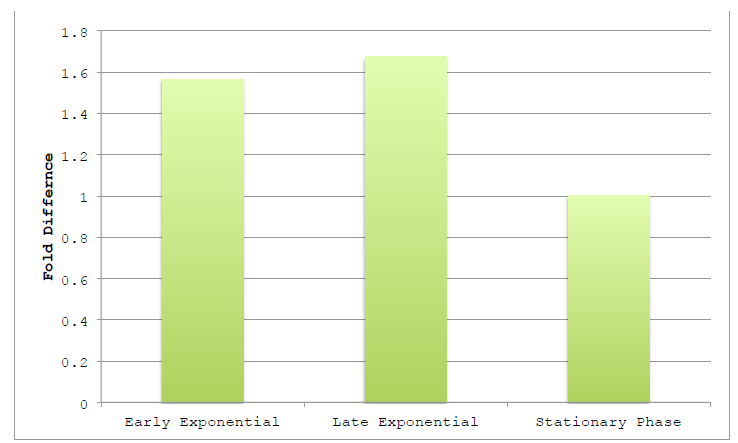
Figure 3.1 Harvested Growth Stage on Assembly Efficiency.
Figure 3.1 Harvested Growth Stage on Assembly Efficiency NEB10β was grown in TB and harvested at various points along the growth curve. Growth phase was judged by optical density at 600 nm, using absorbances of 0.4, 1.0 and >2.0 as markers of early exponential, late exponential and stationary phases, respectively.
Lysates were prepared as previously reported using detergents and two-way assemblies performed. Fold difference represents the change in product band intensity of the sample as compared to the intensity of Stationary Phase product band. Band intensities are calculated relative to the BCP band of the negative control lane.
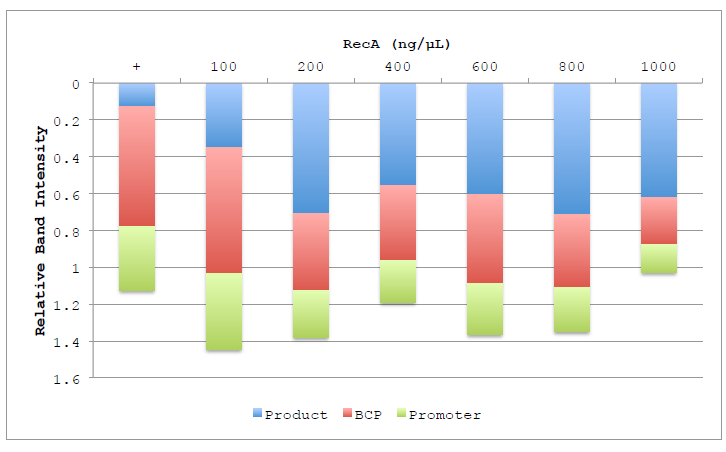
Figure 4.4 Lane composition of RecA titration.
Figure 4.4 Lane composition of RecA titration. Chart shows the relative intensities of the product (Blue), BCP (Red) and Promoter (Green) substrate bands, as well as the low molecular weight band representing degradation products (Purple). Columns are displayed inverted to more closely represent the actual gel and the order in which the bands migrate. Band intensities are calculated relative to the BCP band of the negative control lane.
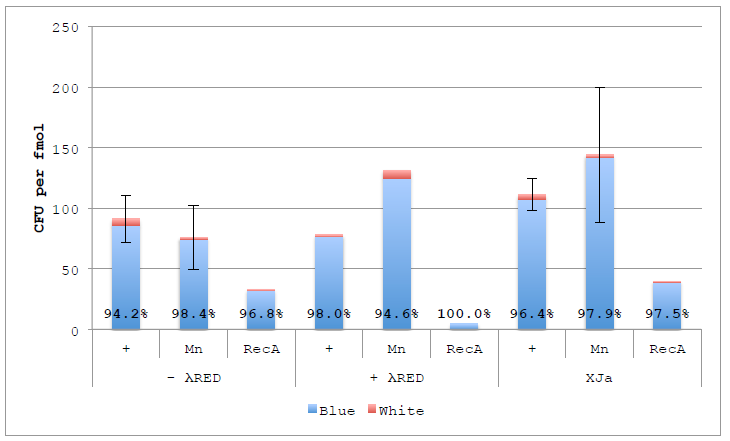
Figure 4.6 ex vivo DNA Assembly with nuc4-.λR and Lambda RED.
Figure 4.6 ex vivo DNA Assembly with nuc4-.λR and Lambda RED. Assemblies were performed using original buffer, supplemented with 1 mM Manganese or 200 ng RecA using autolysed nuc4-, nuc4- with λ Red or XJa. Percentages represent Blue colonies to all colonies (blue and white). Error bars represent standard error of the mean for reactions performed in duplicate (N=2).
EX VIVO PCR AND EX VIVO CLONING
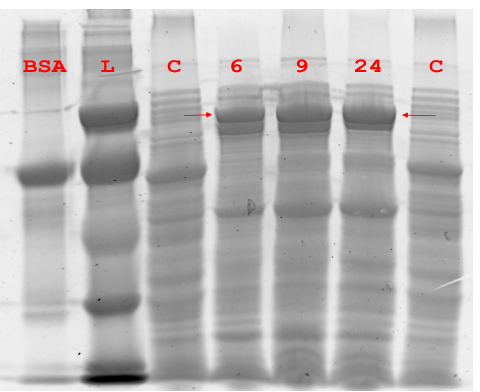
Figure 5.1 Expression of Pfu DNA Polymerase.
Figure 5.1 Expression of Pfu DNA Polymerase. Soluble lysate of NEB10β expressing Pfu was harvested at different time points and separated via denaturing SDS-PAGE. “C” is NEB10β without pUN plasmid, “L” is low molecular weight ladder.
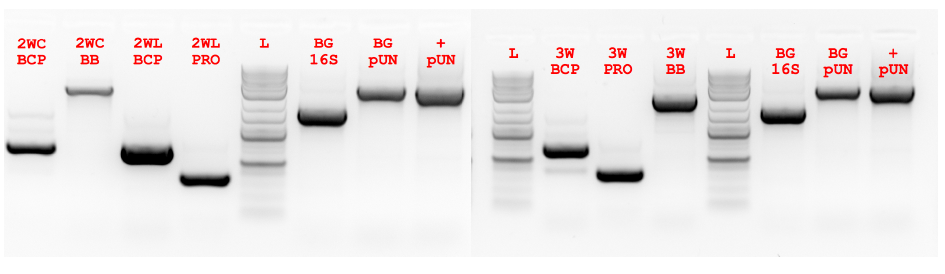
Figure 5.3 ex vivo PCR of 2-way and 3-way Fragments.
Figure 5.3 ex vivo PCR of 2-way and 3-way Fragments. Fragments for 2-way circular (2WC), 2-way linear (2WL) and 3-way circular (3W) were amplified using ex vivo PCR. In addition control PCRs were performed to detect the contribution of background genomic (BG 16S) and pUN-PrhaBAD-Pfu plasmid (pUN) DNA. The positive background (+ pUN) control had purified plasmid template added. There is a clear presence of background amplification and side amplification (see BCP lanes).
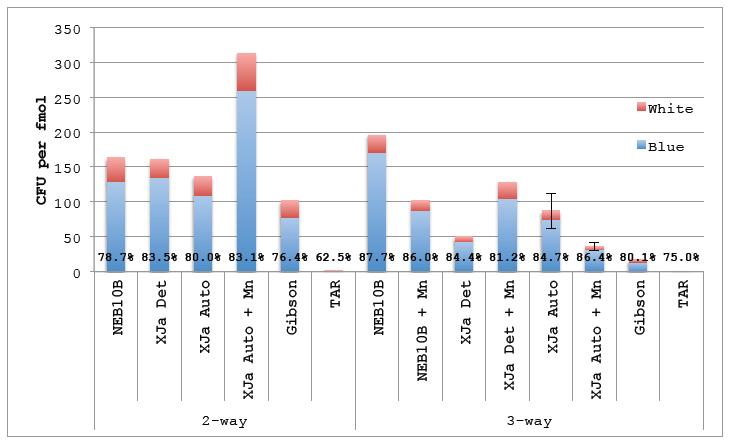
Figure 5.4 ex vivo Cloning Substrate.
Figure 5.4 ex vivo Cloning Substrate. DNA amplified using ex vivo PCR was gel purified and used in ex vivo DNA reactions. 2 μL of the 10 μL reactions were transformed using in-house prepared chemicompetent NEB10β. Gibson lane represents a positive control using Gibson isothermal assembly formulated in-house. TAR is a negative control consisting off DNA fragments transformed directly into NEB10β without assembly reaction. Percentages represent Blue colonies to all colonies (blue and white). Error bars represent standard error of the mean for reactions performed in triplicate (N=3).
CONCLUSION AND FUTURE DIRECTIONS
Responding to the needs of the synthetic biology community for inexpensive, rapid and efficient DNA manipulation we were able to uncover a unique approach to DNA assembly. Working on the observation that all of the enzymes used in prototypical in vitro manipulations of DNA are highly conserved, we incorporated an ex vivo approach to screen several candidate organisms for DNA assembly functions. To accomplish this we designed a very clever assay of DNA assembly.
Our circular assemblies provided a colorimetric output that allowed us to interpret the origins of each resulting transformant. In our intial pilot studies, we used two chloramphenicol plasmids in our two-way assembly and observed the growth of blue (correct), white (background) and red (colonies) – with many background colonies observable. While we realized that this would allow us to track transformation efficiencies by observing fluctuations in background, it was not until subsequent studies did we realize that if the donor plasmids were maintained on different antibiotics than the destination vector (like in the original 3-way assembly; see Figure 2.3), we could track mutational rates.
Because our assemblies join the BCP coding sequence to the destination vector at a crucial junction for proper expression of the BCP protein (across Ribosome Binding Site and Start Codon), there is a low tolerance for error during end joining. Due to the low throughput of performing transformation to assess DNA assembly (~28 hours) we also designed a linear assembly assessed by gel electrophoresis (~3 hours) that greatly expanded our investigative capacities.
MATERIALS AND METHODS
Reagents, DNA and Enzymes
All chemical reagents were purchased from Sigma-Alrich. Divalent metals used in titration experiments were all complexed with chloride (i.e., MgCl2, MnCl2, CaCl2, ZnCl2) and formulated as 1 Molar Stock solutions. All enzymes used in the titration experiment were purchased from NEB, along with the Q5 polymerase used for PCR. All DNA synthesis of primers and synthetic fragments was performed by IDT. The plasmids used for 2-way and 3-way assembly experiments were obtained from the Registry of Standard Biological Parts. Sequences for all DNA used are provided in (DNA Sequences).
Source: Virginia Commonwealth University
Authors: Adam B. Fisher
>> 200+ IoT Led Engineering Projects